Discovery through Partnership | Excellence through Quality
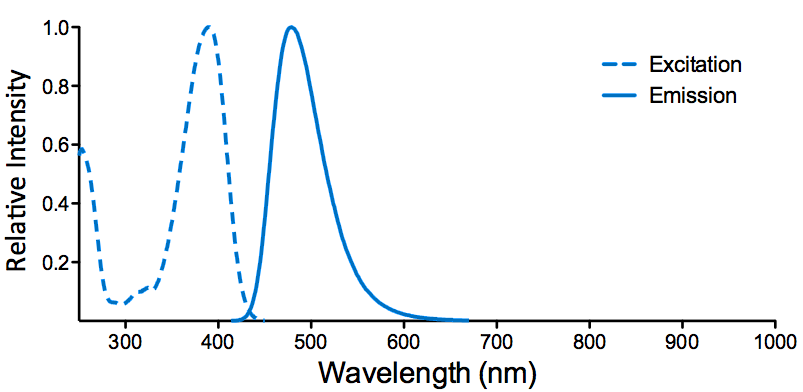
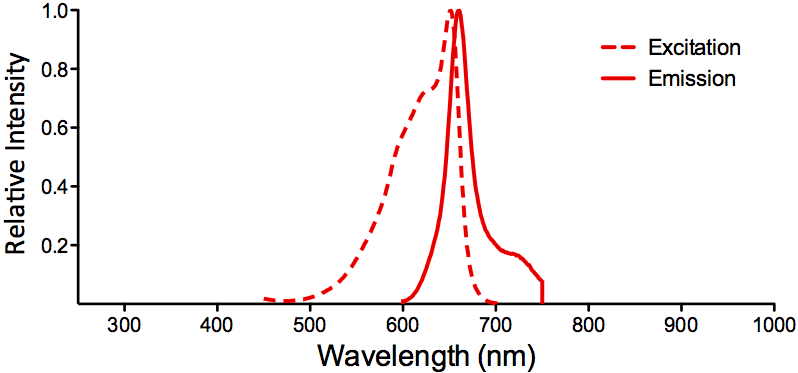
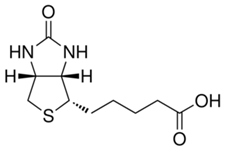
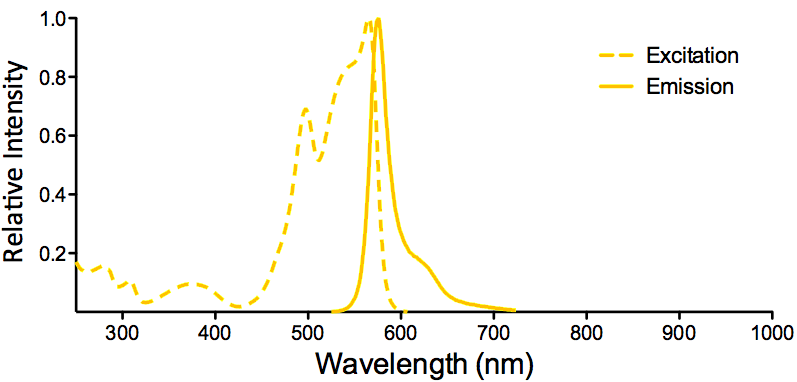
Properties
| Storage Buffer | PBS pH 7.4 |
| Storage Temperature | -80ºC |
| Shipping Temperature | Dry Ice. Shipping note: Product will be shipped separately from other products purchased in the same order. |
| Purification | Ion-exchange Purified |
| Cite This Product | Human Recombinant Transthyretin (TTR) L55P Variant Filaments (StressMarq Biosciences | Victoria, BC CANADA | Catalog# SPR-464) |
| Certificate of Analysis | Certified >95% pure using SDS-PAGE analysis. Low endotoxin <5 EU/mL @ 2mg/mL. |
| Other Relevant Information | For best results, sonicate immediately prior to use. Refer to the Neurodegenerative Protein Handling Instructions on our website, or the product datasheet for further information. Monomer source is catalog# SPR-451. |
Biological Description
| Alternative Names | Transthyretin L55P, TTR L55P, ATTR, Amyloid polyneuropathy, Amyloidosis I, Carpal tunnel syndrome 1, CTS, CTS1, HEL111, HsT2651, PALB, Prealbumin, Prealbumin amyloidosis type I, Prealbumin Thyroxine-binding, TBPA, Thyroxine binding prealbumin, TTHY_HUMAN, TTR |
| Research Areas | ALS Disease, Alzheimer's Disease, Blood, Cardiovascular System, Cell Signaling, Lipid and lipoprotein Metabolism, Metabolism, Neurodegeneration, Neuroscience, Parkinson's Disease, Tangles & Tau |
| Cellular Localization | Cytoplasm, Extracellular exosome, Extracellular Region, Lysosome |
| Accession Number | NP_000362.1 |
| Gene ID | 7276 |
| Swiss Prot | P02766 |
| Scientific Background |
Transthyretin (TTR), encoded by the TTR gene (UniProt ID: P02766), is a tetrameric transport protein primarily responsible for carrying thyroxine and retinol-binding protein in the bloodstream. Under pathological conditions, TTR can misfold and aggregate into amyloid fibrils, contributing to systemic and neurological disorders. The L55P mutation in TTR significantly destabilizes its native tetrameric structure, promoting dissociation into monomers that readily misassemble into amyloid filaments. These filaments are implicated in hereditary transthyretin amyloidosis (ATTRv), a progressive condition marked by polyneuropathy, autonomic dysfunction, and organ impairment. The L55P variant is among the most amyloidogenic mutations, producing highly stable and neurotoxic fibrils that closely resemble those found in patient tissues. TTR L55P filaments serve as a powerful model for studying the molecular basis of amyloid formation, neurotoxicity, and tissue-specific deposition. Their use in experimental systems enables detailed investigation of protein misfolding pathways, cellular stress responses, and neurodegenerative mechanisms. These filaments also facilitate the development and screening of therapeutic agents aimed at stabilizing native TTR, inhibiting fibril formation, or enhancing amyloid clearance. By replicating key features of human amyloid pathology, TTR L55P variant filaments are essential tools in translational research targeting neurodegenerative diseases associated with protein aggregation. |
| References |
1. Zeldenrust S.R., Benson M.D. (2010). Wiley. pp. 795–815. 2. Westermark P., Sletten K., Johansson B., Cornwell G.G. (1990). Proc. Natl. Acad. Sci. U.S.A. 87(7): 2843–5. 3. Andrade C. (1952). Brain. 75(3): 408–27. 4. Coelho T. (1996). Curr. Opin. Neurol. 9(5): 355–9. 5. Jacobson D.R, et. al. (1997). N. Engl. J. Med. 336(7): 466–73. 6. Li X. (2011). Mol Neurodegener. 6(1):79. 7. Lashuel H.A., Wurth C., Woo L., and Kelly J.W. (1999) Biochem. 38(41): 13560-13573. |
Product Images

TEM of Recombinant Human Transthyretin (TTR) L55P Variant Protein Filaments (SPR-464)
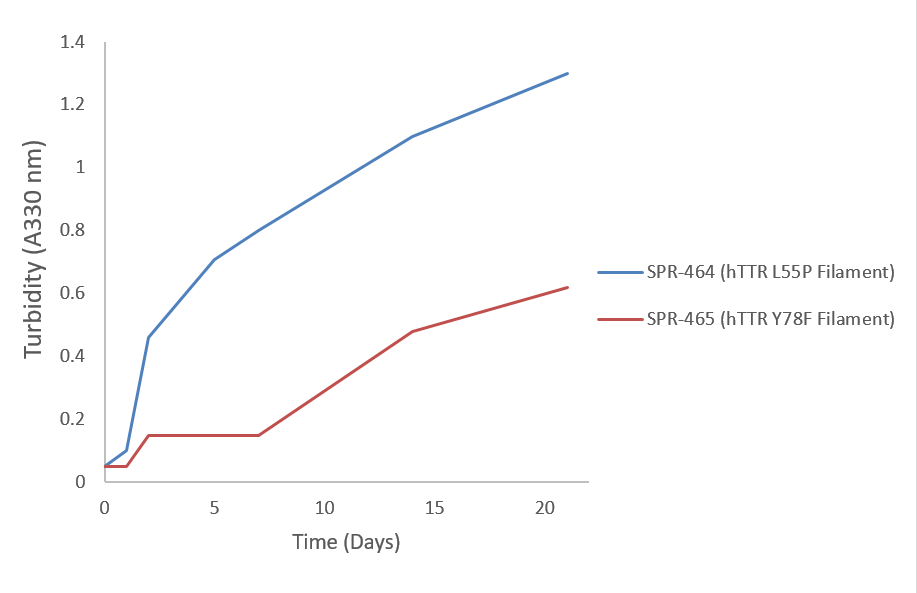
Turbidity Assay shows increased turbidity in Human Recombinant Transthyretin L55P Variant Protein Filaments (SPR-464)
 Powered by Bioz
Powered by Bioz
Reviews
There are no reviews yet.