
TDP-43 & Neurodegeneration: Unraveling Protein Aggregation
First identified in 1995 for its role in repressing HIV-1 gene expression, the transactive response (TAR) DNA-binding protein 43 (TDP-43) has since gained prominence as a key molecular player in the pathogenesis of several major neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Alzheimer’s disease (AD). TDP-43 is a ubiquitously expressed and evolutionarily conserved RNA/DNA-binding protein that plays a fundamental role in multiple aspects of RNA metabolism. These include pre-mRNA splicing, mRNA transport, stability, and translation—processes that are essential for maintaining neuronal integrity and function.
Pathological alterations in TDP-43, such as hyperphosphorylation, ubiquitination, cytoplasmic mislocalization, and aggregation into insoluble inclusions, are now recognized as hallmark features in affected neurons and glial cells across multiple neurodegenerative conditions. These aberrant modifications disrupt its normal nuclear functions and contribute to a cascade of cellular dysfunctions, ultimately leading to progressive neuronal degeneration and cell death.
Although the precise molecular mechanisms underlying TDP-43 proteinopathies remain incompletely understood, ongoing research is increasingly focused on unraveling its role in disease initiation and progression. As such, TDP-43 has emerged as a promising therapeutic target. Strategies aimed at restoring its normal localization, preventing aggregation, or modulating its downstream RNA targets hold potential for developing disease-modifying treatments for ALS, FTD, and other TDP-43-associated neurodegenerative disorders.
What is TDP-43?
TDP-43, encoded by the TARDBP gene, is a ubiquitously expressed nuclear protein that plays a pivotal role in maintaining cellular homeostasis, particularly within neurons. Under physiological conditions, TDP-43 predominantly resides in the nucleus, where it orchestrates a wide array of RNA-related processes. These include alternative splicing, RNA trafficking, regulation of mRNA turnover, and microRNA biogenesis – cellular functions that are essential for proper gene expression and neuronal function. Its ability to shuttle between the nucleus and cytoplasm is tightly regulated through both active transport mechanisms and passive diffusion, allowing it to respond dynamically to cellular signals.
Emerging evidence suggests that TDP-43 is indispensable during early embryonic development, particularly in the formation and differentiation of central nervous system (CNS) cells. Its tightly regulated expression and localization are critical for ensuring proper neuronal development and synaptic connectivity. Consequently, even subtle perturbations in TDP-43 function can have profound effects on neuronal viability and brain homeostasis.
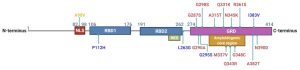
Figure 1. Schematic protein structure of TDP-43, a 414 amino acid protein. The nuclear localization sequence (NLS) is followed by two RNA binding domains (RBD1 & RBD2), a nuclear export sequence (NES), and a glycine rich prion-like (GRD) C-terminus. Mutations reported to increase the risk of ALS (red), FTD (blue) and AD (orange) are indicated. (Figure taken from Meneses et al., 2021 and used under license CC BY 4.0).
The intrinsic propensity of TDP-43 to self-aggregate is largely attributed to its prion-like C-terminal domain, which facilitates aberrant protein-protein interactions and phase separation. During episodes of cellular stress such as oxidative damage, heat shock, or viral infection, TDP-43 can translocate to the cytoplasm and incorporate into stress granules. These transient, membraneless organelles serve as protective units, sequestering stalled mRNAs and RNA-binding proteins to temporarily halt translation and mitigate damage. However, in pathological conditions, these changes promote the formation of insoluble, cytoplasmic aggregates that are a hallmark of TDP-43 proteinopathies.
The Role of TDP-43 in Neurodegeneration
TDP-43 pathology progresses across anatomically connected brain regions, particularly the cortical and limbic systems, through a prion-like mechanism. Misfolded TDP-43 can act as a template, inducing structural changes in native proteins and facilitating the cell-to-cell spread of toxic aggregates. This propagation amplifies neurodegenerative damage and contributes to the clinical variability observed in TDP-43-associated disorders.
In Alzheimer’s disease, TDP-43 pathology is present in 20–57% of cases, rising to 75% in severe forms. These phosphorylated, truncated, and mislocalized cytoplasmic aggregates are most commonly found in limbic regions such as the hippocampus, amygdala, and entorhinal cortex, often alongside hippocampal sclerosis. Their presence is linked to cognitive decline, hippocampal atrophy, synaptic loss, and increased neuroinflammation, which may exacerbate tau and amyloid-β pathology. TDP-43 contributes to AD through both Aβ-dependent and independent mechanisms, and is associated with a distinct clinical phenotype known as LATE (Limbic-predominant Age-related TDP-43 Encephalopathy). Genetic factors such as the APOE4 allele and rare TARDBP mutations may further increase susceptibility.
In ALS and FTD, toxic TDP-43 inclusions within motor neurons and cortical regions are a defining feature, present in approximately 97% of ALS cases. These aggregates—hyperphosphorylated, ubiquitinated, and cleaved—form insoluble deposits that disrupt essential cellular processes and drive neurodegeneration.
StressMarq’s TDP-43 Hexamutant (W→ S) Monomers
A deeper understanding of TDP-43’s aggregation dynamics has opened promising avenues for therapeutic intervention in neurodegenerative diseases such as ALS and FTD. Central to these efforts is the development of tools that allow researchers to study TDP-43’s behavior under controlled conditions. One such tool is StressMarq’s TDP-43 Hexamutant (W→S) Monomers (catalog# SPR-522), engineered with six tryptophan-to-serine mutations (W68S, W113S, W172S, W334S, W385S, W412S) that significantly reduce its aggregation propensity, making it a valuable resource for investigating the molecular mechanisms underlying TDP-43 proteinopathies.
By minimizing spontaneous aggregation, the hexamutant monomers provide a stable baseline for studying how environmental stressors, post-translational modifications, or interacting proteins influence TDP-43 misfolding and pathology. This enables researchers to dissect the early events in TDP-43 dysfunction and to test therapeutic strategies aimed at stabilizing its native conformation or preventing toxic aggregate formation. Furthermore, studies have shown that this hexamutant form has a reduced ability to cross-seed aggregation of other neurodegenerative proteins such as SOD1, offering insights into the interplay between different proteinopathies.
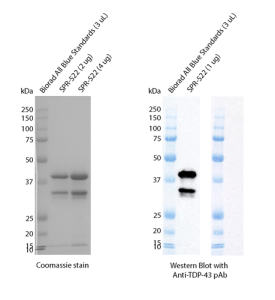
Figure 2. SDS-PAGE analysis of purified TDP-43 Hexamutant Monomers (catalog# SPR-522) (left). Western blot analysis using an anti-TDP-43 antibody (right).
Specialized Tools for Neurodegenerative Disease Research
Alongside strategies that enhance aggregate clearance or modulate TDP-43’s RNA targets, StressMarq’s comprehensive portfolio of neurodegenerative disease proteins is accelerating the development of disease models and drug discovery platforms. Targeting early TDP-43 misfolding, preventing its intercellular spread, and promoting aggregate clearance offer promising therapeutic avenues for ALS, FTD, AD, and related conditions. As a trusted manufacturer of fibrillar and oligomeric protein constructs for in vitro and in vivo research, StressMarq provides a wide range of alpha synuclein, tau, amyloid beta, and SOD1 proteins – available globally through a network of international distributors.
References
-
TDP-43 as a therapeutic target in neurodegenerative diseases: Focusing on motor neuron disease and frontotemporal dementia. Babazadeh, A. et al. Ageing Res Rev. 2023.
-
The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Jo, M. et al. Exp Mol Med. 2020.
-
TDP-43 pathology in Alzheimer’s disease. Meneses, A. et al. Mol Neurodegener. 2021.
- TDP-43 proteinopathy and ALS: Insights into disease mechanisms and therapeutic targets. Scotter, E. L., et al. Neurotherapeutics. 2015.
Leave a Reply