A New Pathway Driving Astrocyte Dysfunction in PD
Parkinson’s disease (PD) is a progressive neurodegenerative disorder marked by the loss of dopaminergic neurons. A defining pathological feature is the accumulation of aggregated alpha synuclein, which forms Lewy bodies within neurons. These insoluble aggregates can be released into the extracellular space, where they seed further pathogenic protein accumulation in neighboring cells. Glial cells, particularly astrocytes, play an active role in clearing extracellular alpha synuclein aggregates. Consequently, their dysfunction may contribute to both the onset and progression of PD.
Previous work by Filippini et al. at the University of Brescia, Italy, has demonstrated that the extracellular chaperone clusterin (Clu) is capable of perturbing the astrocytic uptake of alpha synuclein. The data collected throughout this study establish a robust connection between the LRRK2 G2019S mutation, altered regulation of clusterin, and impaired astrocyte‑mediated uptake of alpha synuclein fibrils. This work is significant because it links a well‑established genetic risk factor for PD to a molecular pathway that shapes glial clearance of toxic protein seeds, offering new insight into mechanisms that may drive disease progression.
Genetic disruption of LRRK2
Current statistics indicate that approximately 10–13% of PD cases can be directly attributed to genetic mutations, while the remaining ~90% are classified as sporadic, arising from a complex interplay of environmental influences and inherent biological susceptibilities. Among the genetic contributors, mutations in the LRRK2 gene, encoding the multidomain scaffolding protein leucine‑rich repeat kinase 2, represent the most common hereditary cause of synucleinopathy. The G2019S variant is particularly notable, as it enhances LRRK2 kinase activity and disrupts endo‑lysosomal and mitochondrial function, ultimately impairing alpha synuclein clearance. Individuals carrying this mutation typically present with late‑onset autosomal dominant Parkinsonism.
To investigate how LRRK2 G2019S affects astrocytic handling of alpha synuclein, the authors isolated primary astrocytes from LRRK2 G2019S knock‑in mice and wild‑type (WT) controls. As an initial validation of their model, they introduced StressMarq’s Mouse Alpha Synuclein Pre‑formed Fibrils (catalog #SPR‑324) to assess fibril uptake. The fibrils were conjugated to a pHrodo fluorescent dye, which increases in brightness upon interaction with acidic environments, facilitating real-time visualization and quantification of fibril internalization through endo‑lysosomal pathways.
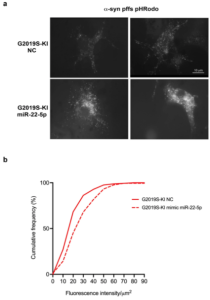
Figure 1. LRRK2 G2019S-KI astrocytes exhibit reduced ability to take up alpha synuclein fibrils. Figure taken from Filippini et al., 2025 used under license CC BY 4.0).
Astrocytes derived from LRRK2 G2019S mice internalized significantly fewer alpha synuclein fibrils compared to WT astrocytes. Moreover, this observation aligns with previous reports demonstrating impaired alpha synuclein clearance in the presence of the G2019S mutation. The reduced uptake suggests compromised astrocytic degradative capacity, which may elevate extracellular alpha synuclein burden and enhance the propagation of pathogenic seeds between neurons.
Astrocyte dysfunction
In order to determine whether reduced uptake of alpha synuclein fibrils in LRRK2 G2019S astrocytes is linked to altered levels of the chaperone clusterin (Clu), the authors performed immunostaining on brain sections isolated from the LRRK2 G2019S and WT mice models. It was found that Clu protein levels were elevated in the mutant brains relative to WT across all detectable forms, including the intracellular precursor (preClu), the cleaved chains, and the extracellularly released protein in conditioned media. Subsequent immunostaining analyses predominantly localized this increase to astrocytes, the primary source of clusterin in the brain.
Clusterin dysregulation
As the discrepancy between Clu mRNA and protein abundance suggested that LRRK2 regulates clusterin post‑transcriptionally rather than at the level of gene expression, Filippini et al. incorporated a puromycin incorporation assay into their workflow. This experiment revealed that the LRRK2 G2019S mutation enhances global protein translation in astrocytes. However, follow‑up assays ruled out involvement of the canonical translational regulator 4E‑BP, indicating that LRRK2 influences protein synthesis through an alternative mechanism.
With the goal of identifying potential post‑transcriptional regulators of clusterin, the authors screened online databases for microRNAs (miRNAs) predicted to target Clu mRNA. Expression profiling in primary astrocytes highlighted miR‑22‑5p as the most consistently genotype‑responsive candidate. Indeed, miR‑22‑5p levels were elevated in LRRK2 knock‑out astrocytes but reduced in LRRK2 G2019S astrocytes relative to WT. A luciferase reporter assay confirmed direct binding between Clu mRNA and a miR‑22‑5p mimic, validating miR‑22‑5p as a bona fide translational regulator of clusterin.
miR‑22‑5p as a regulator of alpha synuclein uptake
Functionally, overexpression of the miR‑22‑5p mimic in primary astrocytes reduced both preClu and cleaved clusterin protein levels without decreasing Clu mRNA abundance, consistent with translational repression. Crucially, restoring miR‑22‑5p levels in LRRK2 G2019S astrocytes increased their uptake of pHrodo‑labeled alpha synuclein fibrils. This was reflected by a rightward shift in cumulative intracellular fluorescence compared to untreated controls, indicating enhanced fibril internalization.
Together, these data support a mechanistic model in which dysregulated miR‑22‑5p contributes to elevated clusterin levels in LRRK2 G2019S astrocytes, thereby limiting their capacity to internalize alpha synuclein fibrils. Normalizing miR‑22‑5p expression reverses this effect by lowering clusterin abundance and improving astrocytic clearance of extracellular fibrils, linking miRNA‑mediated translational control directly to glial processing of pathogenic alpha synuclein seeds.
Summary
Filippini et al. present a mechanistic LRRK2-miR‑22‑5p-clusterin pathway that regulates astrocyte uptake of pathogenic alpha synuclein fibrils. In this model, the LRRK2 G2019S mutation boosts global protein translation while suppressing miR‑22‑5p. Reduced miR‑22‑5p lifts translational repression on clusterin, elevating clusterin protein levels in astrocytes. This increase limits fibril internalization and impairs clearance of extracellular alpha synuclein seeds.
Reintroducing miR‑22‑5p normalized clusterin levels and restored fibril uptake in G2019S astrocytes, demonstrating a direct functional link between this miRNA and astrocytic proteostasis. These findings show how a major genetic risk factor for Parkinson’s disease disrupts protein‑handling pathways in ways that may amplify extracellular fibril burden and promote pathological spread.
Taken together, the study positions the LRRK2-clusterin axis as a promising therapeutic target for enhancing astrocytic clearance of alpha synuclein. Modulating miR‑22‑5p or directly targeting clusterin could potentially normalize uptake mechanisms in individuals carrying PD‑associated LRRK2 mutations. Future work assessing the impact of LRRK2 kinase inhibitors, miRNA‑based therapeutics, or other approaches that modulate this pathway will be crucial in determining whether these strategies can reduce the spread of alpha synuclein pathology and ultimately alter disease progression.
Related StressMarq products
StressMarq Biosciences manufactures a diverse portfolio of alpha synuclein, tau, amyloid beta, and TDP-43 proteins designed to support advanced neuroscience research. These include an innovative range of oligomeric, fibrillar and monomeric protein preparations, alongside antibodies, kits, and small molecules. Visit our website for more information, including the latest scientific publications using our specialized protein constructs.
References
- Astrocytes carrying LRRK2 G2019S exhibit increased levels of clusterin chaperone via miR-22-5p and reduced ability to take up α-synuclein fibrils. Filippini, A. et al. Acta Neuropathol Commun. 2025.
Leave a Reply