
Halting the Progression of Alzheimer’s Disease
Alzheimer’s disease (AD) is a highly prevalent neurodegenerative disorder, leading to a gradual decline in memory, cognition, and behavioural capacity. Although AD primarily affects individuals later in life, the early-onset form of the disease results in patients displaying cognitive decline in their 40s. In all cases, the primum movens for the development of AD is the accumulation of amyloid beta plaques. Buildup begins decades before symptoms of cognitive decline may begin to affect day-to-day life. Moreover, disease progression triggers the conversion of a normally expressed neuronal protein — tau — into an unregulated toxic state, with both proteins working in tandem to activate destructive biochemical pathways. This complex process ultimately results in irreversible neuronal cell damage.
Understanding the pathological relationship between amyloid beta and tau is of great importance to researchers investigating AD. A new study published in Cell by Chen et al., describes how a mutation in a gene linked to lipid transport may have an innate protective effect against AD. This is proposed to be achieved through, among other factors, the enhancement of tau uptake and degradation by microglial immune cells. The cellular and molecular mechanisms elucidated in this study may contribute to the identification of new therapeutic targets and strategies for AD.
Employing a mouse model to understand early-onset AD
The early-onset, familial form of AD arises from defects in one of three genes: PSEN1, PSEN2 or APP. Indeed, alterations in any one of these genes stimulates the toxic accumulation of amyloid beta protein in the brain. One such mutation is found in the Presenilin 1 gene — PSEN1-E280A — involved in amyloid beta processing and generation. In 2019, a case study published in Nature Medicine described a woman from a Colombian family carrying this mutation, who, unlike her relatives, was able to circumvent the development of early-onset AD for 30 years. Despite demonstrating unusually high levels of amyloid beta, her brain exhibited limited toxic tau pathology and minimal indicators of neurodegeneration. It was proposed that this mitigation of early-onset AD was due to the woman being a carrier of two copies of the rare APOE3 Christchurch (R136S) mutation.
To study the protective effects of the APOE3ch mutation against AD, researchers at the Washington University School of Medicine in St. Louis employed an AD mouse model carrying mutant forms of PSEN1 and amyloid beta. The goal of this model was to recreate the progressive buildup of amyloid beta deposits observed in AD patient brains. Injection of insoluble tau fibrils isolated from brain extracts of AD patients was then used to mirror advanced-stage AD, whereby tau acted to induce further protein misfolding and aggregation.
Given that the APOE3 gene is involved in lipid transport, researchers initially sought to assess whether the Christchurch variant affected this process. Similar to the case involving the Colombian woman, the mice displayed abnormally high levels of lipids in the peripheral nervous system (PNS) but not in the central nervous system (CNS). Furthermore, as APOE3 has previously been linked to neuroinflammation, this parameter was also the subject of preliminary investigation. In this regard, the Christchurch variant was shown to have protective effects against neurodegenerative disease progression — curtailing amyloid beta buildup in plaques, and tau spreading and aggregation surrounding the amyloid plaques themselves.
Enhanced phagocytosis of tau by microglia
Amyloid beta plaques are known to attract microglial cells, which are thought to act in the clearance of amyloid plaques in AD. Abnormalities in microglial activation are often exhibited in patients experiencing symptoms of dementia. Analysing the activity of amyloid plaques subjected to the effects of the Christchurch variant revealed enhanced microglial recruitment. To elucidate the effect of this variant in the cellular processing of tau by microglial cells, StressMarq’s Human Tau-441 (2N4R) P301S Mutant PFFs (catalog# SPR-329) were utilized.
StressMarq’s recombinant tau pre-formed fibrils were fluorescently labelled to monitor the phagocytic process. Conjugation to a pH-sensitive fluorescent dye indicated that tau was internalized into the acidic lysosomal compartments of microglia during phagocytosis. Interestingly, the APOE3ch mutation resulted in increased phagocytosis and degradation of the human tau fibrils due to destabilized binding to heparan sulphate proteoglycan cell surface receptors. Further adding to its protective effect, the Christchurch variant was able to suppress the ability of microglial to release tau seeding and spreading, thus reducing further misfolding and aggregation of tau.
Examining cellular mechanisms of AD using StressMarq products
Although rare, familial AD can affect individuals much earlier on in life. Afflicted patients commonly begin to demonstrate symptoms of cognitive decline in their 40s, although individuals may be diagnosed as early as their 20s. Health complications attributed to AD significantly increase the risk of premature death. Understanding the factors affecting how AD progresses from the initial build-up of amyloid beta in plaques before synergizing with tau hyperphosphorylation and consequent decline in cognitive function remains ill-defined. By altering the molecular interactions influencing the phagocytosis of tau aggregates, the APOE3 Christchurch mutation halts the progression of AD. StressMarq’s tau fibrils were instrumental in deciphering how microglial cells with the Christchurch variant enhanced the degradation of tau aggregates. Mimicking the effects of the Christchurch variant may now assist in developing therapeutics to prevent the progression of AD.
By altering the molecular interactions influencing the phagocytosis of tau aggregates, the APOE3 Christchurch mutation appears to protect against the progression of AD. StressMarq’s Tau-441 (2N4R) P301S Mutant PFFs were instrumental in deciphering how microglial cells carrying the Christchurch variant enhanced the degradation of tau aggregates. Mimicking the effects of APOE3ch could be a valuable tool in the pursuit of therapeutic treatments for Alzheimer’s disease.
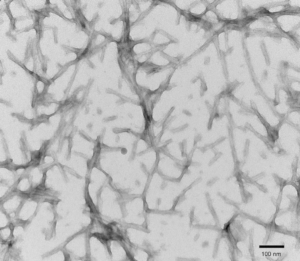
Figure 1. TEM of Tau-441 (2N4R) P301S Mutant (PFFs) at 150kx magnification. HV = 80kV. Fibrils were sonicated and stained with uranyl acetate (catalog# SPR-329)
Related StressMarq products
StressMarq manufactures a range of proteins for neurodegenerative disease research, such as oligomeric amyloid beta preparations and tau pre-formed fibrils and monomers – suitable for molecular, cellular and animal research models. The newly released Tau dGAE (297-391) AD-mimic PFFs, (catalog# SPR-502), replicate the disease-specific tau fold found in individuals with Alzheimer’s disease to mimic AD pathology in research models.
Supplemental reading material
- Clues to preventing Alzheimer’s come from patient who, despite genetics, evaded disease. Source: WUSTL Medicine
- Alzheimer’s disease evading patient offers clues on breaking amyloid, tau link. Source: Genetic Engineering & Biotechnology News
- Rare gene mutation shields woman from Alzheimer’s. Source: Neuroscience News
References
- APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread. Chen, Y. et al. Cell. 2023.
- Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Arboleda-Velasquez, J. F, et al., Nature Medicine. 2019.
Leave a Reply