
Mitophagy in Mitochondrial Diseases
The word mitophagy derives from the Greek ‘phageîn’ for ‘cell eating’ and describes the selective clearance of damaged mitochondria. Mitophagy occurs in cells exposed to damage or stress, such as oxygen deprivation (hypoxia). The process acts as a quality control step limiting the accumulation of mutations in and subsequent release of the mitochondrial DNA.
Mitophagy is a hallmark of age-related mitochondrial pathology in mammalian muscle. A growing number of reports show that mitochondrial function is disrupted in neurodegenerative disorders, aging, cardiovascular disease, and cancer. Common symptoms of these diseases are muscle weakness and fatigue.
Mitophagy and Parkinson’s disease
Parkinson’s disease is a neurodegenerative disorder pathologically characterized by death of the dopamine-producing neurons in the substantia nigra. There are several genetic mutations implicated in Parkinson’s disease, including loss of function PINK11 and Parkin2. Loss of function in either of these genes results in the accumulation of damaged mitochondria, and aggregation of proteins or inclusion bodies – eventually leading to neuronal death.
Mitochondria dysfunction is thought to be involved in Parkinson’s disease pathogenesis. In non-genetically linked Parkinson’s disease, the disease is commonly caused by dysfunctional mitochondria, cellular oxidative stress, autophagic alterations and the aggregation of proteins. These can lead to mitochondrial swelling and depolarization. It is important to keep the dysfunctional mitochondria regulated, because all of these traits could be induced by mitochondrial dysfunction that can lead to cell death3. Disorders in energy creation by mitochondria can cause cellular degeneration, like those seen in the substantia nigra4.
Mitochondrial disease progression
A study published in Cell Metabolism has now revealed that mitophagy disruption is critical in mitochondrial muscle disease. To study the effect of mitophagy on muscle disease, the researchers used a mouse model for premature aging. In this, the muscle cells display deficiencies in normal mitochondrial functions. The muscle fibers of this mouse model represent the different stages of
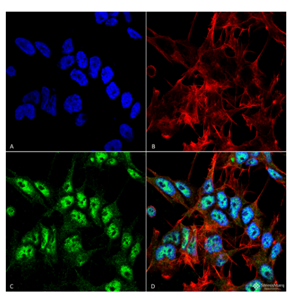
Figure 1. Immunofluorescence analysis using Rabbit Anti-Beclin 1 Polyclonal Antibody (catalog# SPC-600) in Human Neuroblastoma cell line (SK-N-BE).
mitochondrial disease progression that occur in patients and the elderly5.
In the early phase of muscle disease, nuclei typically positioned in the periphery of the muscle fibers can be found clustered in the center of cells. In the later stages of muscle disease, ragged-red fibers (RRFs) are present. RRFs are named as such because as muscle disease progresses, mitochondrial aggregation causes the contours of muscle cells to become irregular. The diseased muscle fibers have a “ragged” appearance under the microscope when using the Gomori trichrome, a red stain.
In the study, the authors showed that mitophagy is increased in the early stages of muscle disease. As muscle disease progresses, mitophagy is stalled, which activates the mTORC1 signaling pathway.
The researchers identified a potential therapy – the mTORC1 inhibitor, rapamycin. Rapamycin-treated mice resulted in the restoration of the levels of mitophagy in muscle cells displaying advanced disease.
Factors behind altered mitophagy
To study the molecular factors behind altered mitophagy in mitochondrial diseases, the researchers used StressMarq’s Beclin 1 antibody (catalog# SPC-600). Beclin 1 is a key factor in both general autophagy and mitophagy. In autophagy it has a central role, being responsible for the initial formation of the double membrane that forms the autophagosome. In mitophagy, Beclin 1 is thought to be important in targeting mitochondria into autophagosomes.
Beclin 1 was more pronounced in RRFs fibers from patients with mitochondrial DNA changes. Yet, the problem was shown to be complex, as Beclin 1 and increased lysosomal amounts were present but mitophagy was stalled.
In summary, the researchers delineate the complex situation in muscles associated with pathological conditions such as aging and mitochondrial disease. The study authors describe how disease muscle cells have either elevated or deficient mitophagy. This drives cell-autonomous regulatory mechanisms for mitochondrial turnover. The study proposes therapeutic solutions that activate mitophagy in mitochondrial diseases affecting muscles, such as aging-related muscle wasting.
Related StressMarq Products
Specializing in products for neurodegenerative research, StressMarq offers a range of antibodies, proteins, kits and small molecules for life science research. The extensive list includes another Beclin 1 Antibody (catalog# SPC-601) and various Beclin 2 Antibodies (catalog# SPC-602, 603, 604). Also, relevant to mitophagy, StressMarq offers a Rubicon Antibody (catalog# SPC-668) and PINK1 Antibody (catalog# SMC-450).
References
- Hereditary early-onset Parkinson’s disease caused by mutations in PINK1, Valente, E. et al., Science. 2004; 304 (5674): 1158–60.
- Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism, Kitada, T. et al., Nature. 1998; 392 (6676): 605–8.
- Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD, Esteves, A., Arduíno, D., Silva, D., Oliveira, C., Cardoso, S., Parkinson’s Disease. 2011; 693761.
- Mitochondrial fusion/fission, transport and autophagy in Parkinson’s disease: when mitochondria get nasty, Arduíno, D., Esteves, A., Cardoso, S., Parkinson’s Disease. 2011; 767230.
- Mosaic dysfunction of mitophagy in mitochondrial muscle disease, Mito, T. et al., Cell Metabolism. 2022; 34(2).
Leave a Reply