
The Pathogenesis of Multiple System Atrophy (MSA)
Multiple System Atrophy (MSA) belongs to the family of amyloid-related diseases, most of which manifest in neurodegeneration. MSA is defined by the misfolding and accumulation of amyloid alpha synuclein deposits in oligodendrocytes, to form Glial Cytoplasmic Inclusions (GCIs) leading to a rapidly progressive neurodegenerative disease1.
Pathology
Oligodendrocytes are the myelinating glia of the central nervous system. The identification of alpha synuclein in the GCIs of MSA patients’ oligodendrocytes 2, 3 was complemented with the identification of alpha synuclein accumulation in the Lewy Bodies present in patients suffering from Parkinson’s disease with Dementia (PDD) or Dementia with Lewy Bodies (DLB)2. These discoveries led to the classification of PD, PDD, DLB, and MSA as Synucleinopathies4. Yet, MSA as a Synucleinopathy, is different from Parkinson’s disease (PD) and Dementia of Lewy Bodies (DLB) in which the amyloid alpha synuclein deposits emerge in neurons instead.
Amyloid Fibrils and Oligomers
Fibrils typically result from the assembly of several protofilaments, each made up of a double layer of β-sheets. Oligomeric species accumulate as intermediates in the process of fibril formation, are reported to be highly cytotoxic,5, 6, 7 and they may be key players in the initiation of disease and in its spreading through cell-to-cell transmission8.

Figure 1: Three-dimensional reconstructions of the two main size subgroups of oligomers of purified oligomeric samples. Reproduced from Chen et al., Structural characterization of toxic oligomers that are kinetically trapped during α-alpha synuclein fibril formation, PNAS, 2015. 112(16):1994-2003.
Cytotoxicity of Alpha Synuclein
The discovery that alpha synuclein is the main constituent of LBs and GCIs led to several studies investigating the molecular mechanisms underlying the pathogenesis of PD and MSA.
Aggregation
In one study, human embryonic kidney (HEK) cell lines and SH-SY5Y neuroblastoma cell lines, engineered to express alpha synuclein fused to either DsRed or Green Fluorescent Protein (GFP) were co-cultured, each expressing the other fusion protein. Regardless of cell type used, the investigators found that GFP-positive alpha synuclein had propagated to cells expressing alpha synuclein fused to DsRed, and vice versa9. In HEK cells overexpressing wild-type alpha synuclein, exogenous alpha synuclein preformed fibrils (PFFs) could induce intracellular aggregation10. Alpha synuclein PFFs could also induce endogenous alpha synuclein aggregation in primary neuron cultures, leading to synaptic dysfunction and neuron death11.
Prion Behavior
The fragments of amyloid fibrils, so-called “seeds” or “pre-formed fibrils” of alpha synuclein, when injected into the cells or the brains of mice, could propagate and be transmitted. A single intra-striatal inoculation of synthetic alpha synuclein fibrils in wild-type non-transgenic mice led to cell-to-cell transmission of pathologic alpha synuclein and Parkinson’s-like Lewy pathology in anatomically interconnected regions. This pathology led to progressive loss of dopamine neurons in the substantia nigra pars compacta, but not in the adjacent ventral tegmental area (VTA) and was accompanied by reduced dopamine levels culminating in motor deficits10. Intracerebral injections of sarkosyl-insoluble alpha synuclein from brains of DLB patients induced hyperphosphorylated alpha synuclein pathology in wild-type mice. Injecting fibrils of recombinant human and mouse alpha synuclein also efficiently induced similar pathologies in wild-type mice. Endogenous mouse alpha synuclein started to accumulate 3 months after inoculation, while the injected human alpha synuclein fibrils disappeared in about a week12. These were indications of prion-like behavior.
Alpha synuclein expression levels decrease during oligodendrocyte maturation13 making it credible that external alpha synuclein contribute to GCI deposits. Thus, the internalizing of alpha synuclein during pathogenesis was investigated and confirmed14. The concept of self-propagation by templating misfolding, was first described for prions as disease-causing agent for scrapie (in sheep) and Creutzfeldt–Jakob disease15 but it is suggested that the concept also applies to Alzheimer’s disease and Parkinson’s disease16. Can MSA be added to the list?
To confirm the principle of prion-behavior of the MSA patient-derived aggregates, the mutated a-syn140*A53T–YFP cell line was infected with the aggregates of one MSA patient (MSA14), and stably propagating cells were sorted by FACS to establish two monoclonal cell lines (MSA14-11 and MSA14-M1). Lysates from these two cell lines were used to infect a naïve mutated cell line and compared with the effects of lysates from uninfected cells. MSA14-11 and MSA14-M1 lysates both induced a robust infection compared to the uninfected lysates17.
Pathology
Giasson et al.18 developed a transgenic mouse model expressing human alpha synuclein with the A53T mutation expressed under the mouse prion protein, Prnp, promoter. The homozygous mice spontaneously developed motor deficits and substantial phosphorylated alpha synuclein pathology. In contrast, the hemizygous mice did not spontaneously develop these symptoms but did when inoculated with brain homogenate prepared from two MSA patient samples19.
Sarkosyl-insoluble fractions from MSA brain extracts also induced cerebral pSer129 alpha synuclein deposition 6–9 months after intracerebral inoculation of transgenic mice expressing wildtype human alpha synuclein under its own promoter20. Inoculation with brain homogenate prepared from PD patient samples did not transmit neurological disease to the hemizygous mice, suggesting that the two synucleinopathies arise from distinct conformations of misfolded alpha synuclein21.
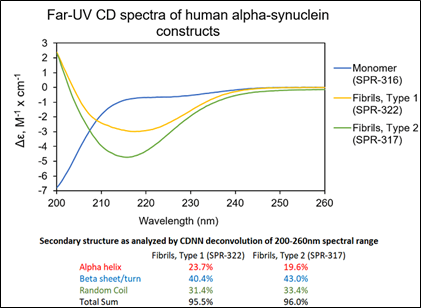
Figure 2: UV-CD data suggests StressMarq’s Alpha Synuclein Pre-formed Fibrils (PFF), Type 1 (catalog# SPR-322) and Type 2 (catalog# SPR-317) both have a high beta sheet/turn content, yet do have small secondary structure differences.
StressMarq Products for MSA Research
Specializing in research tools for studying neurodegenerative diseases, StressMarq offers a wide variety of Alpha Synuclein Monomers, Oligomers and Pre-formed fibrils, useful for modelling synucleinopathies because of their ability to induce aggregation and pathology in different cellular and animal models. These include Kinetically Stable Alpha Synuclein Oligomers (catalog# SPR-484), developed without any inducers or inhibitors, which are toxic to dopaminergic neurons and induce phosphorylation of alpha synuclein pSer129.
In addition to alpha synuclein fibrillar proteins, StressMarq offers a range of antibodies, proteins, kits, and small molecules for MSA and Parkinson’s disease research. Also available are a range of products to study amyloid beta and tau, which are primarily associated with Alzheimer’s disease research.
References
- Linard, M. et al. Infectious Agents as Potential Drivers of α-Synucleinopathies. Mov Disord. 2022 Jan 18.
- Spillantini, MG. et al. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998 Jul 31;251(3):205-8.
- Wakabayashi, K. et al. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998 Jun 19;249(2-3):180-2.
- Hardy, J. & Gwinn-Hardy, K. Genetic classification of primary neurodegenerative disease. Science. 1998 Nov 6;282(5391):1075-9.
- Chiti, F. & Dobson, CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333-66.
- Bucciantini, M. et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002 Apr 4;416(6880):507-11.
- Stefani, M. & Dobson, CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl). 2003 Nov;81(11):678-99.
- Nath, S. et al. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of β-amyloid. J Neurosci. 2012 Jun 27;32(26):8767-77.
- Hansen, C. et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011 Feb;121(2):715-25.
- Luk, KC. et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009 Nov 24;106(47):20051-6.
- Volpicelli-Daley, LA. et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011 Oct 6;72(1):57-71.
- Masuda-Suzukake, M. et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013 Apr;136(Pt 4):1128-38.
- Djelloul, M. et al. Alpha-Synuclein Expression in the Oligodendrocyte Lineage: an In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell Reports. 2015 Aug 11;5(2):174-84.
- Reyes, JF. et al. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia. 2014 Mar;62(3):387-98.
- Prusiner, SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982 Apr 9;216(4542):136-44.
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science. 2015 Aug 7;349(6248):1255555.
- Woerman, AL. et al. α-Synuclein: Multiple System Atrophy Prions. Cold Spring Harb Perspect Med. 2018 Jul 2;8(7):a024588.
- Giasson, BI. et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002 May 16;34(4):521-33.
- Watts, JC. et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013 Nov 26;110(48):19555-60.
- Bernis, ME. et al. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun. 2015 Nov 26;3:75.
- Prusiner, SB. et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015 Sep 22;112(38):E5308-17.
Leave a Reply