
Transthyretin Amyloid Fibrils in Amyloidosis Research
Protein misfolding and abnormal aggregation are central issues in several neurodegenerative disorders as well as amyloid diseases. Amyloidosis is a group of diseases caused by the accumulation and deposition of amyloid fibrils. There are many types of amyloidosis. Amyloid fibrils have drawn the attention of many researchers attempting to understand disease mechanisms and discover effective treatments1.
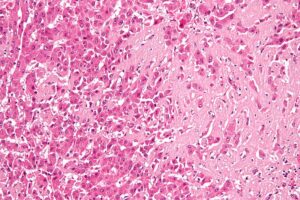
High magnification micrograph of hepatic amyloidosis, also amyloidosis of the liver. Nephron / CC BY-SA (https://creativecommons.org/licenses/by-sa/3.0)
What is Transthyretin Amyloidosis?
Transthyretin (TTR) is a 55kDa homotetrameric protein in the serum and cerebrospinal fluid that transports thyroxine and retinol. Under pathological conditions the tetramer aggregates into insoluble amyloid fibrils which are deposited in the heart and nervous system2.
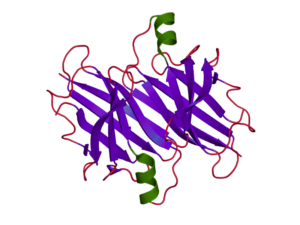
Transthyretin protein structure
Transthyretin amyloidosis (ATTR) is a rare, complex and slow-progressing disease. It occurs when transthyretin misfolds and aggregates into amyloid deposits in the heart, nerves and other organs of the body leading to permanent damage and death3,4. Liver transplantation was the first therapeutic approach for patients with hereditary ATTR which showed to replace mutant-TTR with wild type -TTR and increase patient survival5. However, the limitations of this procedure led to the discovery of new therapies.
In recent years several therapeutic strategies have been proposed such as stabilizing TTR tetramers using small molecules (tafamidis and diflunisal)6,7. There is also increasing interest in gene-silencing therapies that inhibit WT- TTR and mutated TTR synthesis in the liver (patisiran and inotersen)7. These novel therapies are very promising in preventing the progression of Transthyretin Amyloidosis.
Types of Transthyretin Amyloidosis
Wild Type Transthyretin Amyloidosis :
- Senile Systemic Amyloidosis (SSA) is a late-onset non-hereditary form caused by the aggregation of the wild type Transthyretin (WT- TTR) leading to heart failure8.
Hereditary Transthyretin Amyloidosis:
- Familial Amyloid Polyneuropathy (FAP) is a rare autosomal dominant condition caused by the accumulation of TTR amyloid fibrils in the peripheral nervous system. Mutations of the TTR gene destabilize the protein, causing it to abnormally aggregate9.
- Familial Amyloid Cardiomyopathy (FAC) is also associated with various mutations and caused by the accumulation of TTR in the heart3.
The role of TTR amyloid fibrils in research
Understanding the mechanisms of how a folded protein converts into insoluble amyloid fibrils is the most critical step in developing inhibition strategies against amyloidosis10. In neurodegeneration research amyloid seeding seems to be a mechanism of spreading pathology. TTR preformed fibrils can be injected into mice where they act as seeds that induce pathology. It is known that amyloid seeding is accelerated by the presence of preformed fibrils and leads to pathology spreading.
In 2018, a research group from UCLA used fibrils extracted from human cardiac tissue to study ATTR fibril formation11. They found that the ex vivo amyloid fibrils can seed fibril formation of both wild-type and mutant TTR in vitro and proposed that inhibition of amyloid seeding could be a promising therapeutic strategy for ATTR. Tetramer stabilizers like tafamidis and diflunisal could only inhibit protein aggregation in the absence of seeds but they were ineffective when seeds were present12,13.
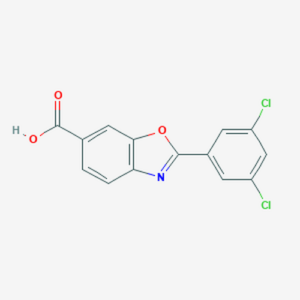
Chemical structure of Tafamidis, a TTR kinetic stabilizer.
The same group, in 2019, found that inhibitor peptides TabFH2 can bind ex-vivo fibrils and efficiently inhibit amyloid seeding. These peptide inhibitors were originally designed and optimized to cap the tip of TTR fibrils by binding β-strands F and H14.
Models of Transthyretin Amyloidosis
Disease models are crucial in understanding the mechanisms of ATTR amyloidosis pathogenesis and testing novel therapeutic strategies. Creating an ideal model that can display the pathophysiology of ATTR has become very challenging as the disease is very complex, genetically heterogeneous and affects multiple organs. Several animal models have been used such as the invertebrates Drosophila melanogaster and Caenorhabditis elegans, rodents and non-human primates as well as cellular models (iPSCs)15.
Transgenic mice expressing WT-TTR or mutant TTR gene constructs have been used for years in ATTR research with no success in reproducing the disease15,16. Two different research groups in 2004 injected ex-vivo ATTR preformed fibrils into transgenic mice to test amyloid seeding but they did not observe acceleration of TTR deposition17,18. Recently, two humanized mouse models showed TTR deposition in the peripheral nerves but they could not reproduce all the features of the disease. Several research groups are working to develop new efficient disease models that can recapitulate TTR amyloidosis19,20.
Transthyretin Mutations
More than 100 different mutations in the TTR gene are associated with hereditary ATTR amyloidosis. They can destabilize and dissociate the tetrameric structure of TTR and cause extracellular deposition of amyloid fibrils. The TTR- V30M variant is the most prevalent mutation associated with familial amyloid polyneuropathy and affects peripheral and autonomic nerves. TTR- L55P variant is the most amyloidogenic. Experiments have shown that L55P-TTR slowly dissociates to folded amyloidogenic monomer that self-assembles into protofilaments under physiological conditions. Furthermore, this variant seems to be more sensitive in acidic conditions. The WT-TTR tetramer shows no dissociation to monomer under normal conditions21,22.
The variant Y78F – TTR has been shown to expose a cryptic epitope which is recognized by a monoclonal antibody that reacts only with highly amyloidogenic mutants. A previous study showed that immunization of transgenic (TTR- V30M) mice with the TTR-Y78F variant, reduced human TTR deposition and cleared TTR amyloid deposits in the gastrointestinal tract. The immunotherapy with the human TTR – Y78F seems to be a promising therapeutic tool for TTR amyloidosis23.
StressMarq offers L55P and Y78F mutant TTR monomers and filaments for researchers to study the mechanisms of TTR aggregation as well as to discover novel therapeutic strategies.

TEM of Human Recombinant Transthyretin L55P Variant Protein Filaments (catalog# SPR-464).
REFERENCES
- Amyloid fibrils: abnormal protein assembly. Rambaran RN et al. (2008) Prion 2(3):112‐
- Uncovering the Mechanism of Aggregation of Human Transthyretin. Saelices L. et al. (2015) J Biol Chem 290(48):28932‐
- Transthyretin (TTR) cardiac amyloidosis. Ruberg FL. et al. (2012) Circulation. 126(10):1286‐
- Transthyretin: the servant of many masters. Buxbaum J.N. et al. (2009). Mol. Life Sci. 66: 3095–3101.
- Liver transplantation and transthyretin amyloidosis. Benson MD.(2013) Muscle Nerve 47(2):157-62.
- TTR (Transthyretin) stabilizers are associated with improved survival in patients with TTR Cardiac Amyloidosis. Rosenblum, H., et al. (2018) Circ Heart Fail. 11(4): e004769.
- Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care. Luigetti M. et al. (2020) Ther Clin Risk Manag. 16:109‐
- Fibril in senile systemic amyloidosis is derived from normal transthyretin. Westermark P. et al. (1990) Proc Natl Acad Sci U S A 87, 2843-2845
- Familial Amyloid Polyneuropathy. Çakar A. et al. (2019) Noro Psikiyatr Ars. 56(2):150–156.
- Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms. Giorgetti S. et al. (2018) Int J Mol Sci.19(9):2677.
- Amyloid seeding of transthyretin by ex vivo cardiac fibrils and its inhibition. Saelices L. et al. (2018) Proc Natl Acad Sci U S A.115(29): E6741-E6750.
- Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. Adamski-Werner SL. et al. (2004) J Med Chem 47:355–374.
- Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Bulawa CE, et al. (2012) Proc Natl Acad Sci USA 109:9629–9634.
- A pair of peptides inhibits seeding of the hormone transporter transthyretin into amyloid fibrils. Saelices L. et al. (2019) J. Biol. Chem. 294(15):6130-6141.
- Contributions of Animal Models to the Mechanisms and Therapies of Transthyretin Amyloidosis. Ridwan B.I. et al. (2019) Front. Physiol. 10:338.
- Animal models of human amyloidosis: are transgenic mice worth the time and trouble? Buxbaum J. N. (2009). FEBS Lett. 583: 2663–2673.
- Amyloidogenesis is neither accelerated nor enhanced by injections of preformed fibrils in mice transgenic for wild-type human transthyretin: The question of infectivity. Tagoe C.E. et al. (2004) Amyloid 11:21–26.
- Deposition of transthyretin amyloid is not accelerated by the same amyloid in vivo. Wei L. et al. (2004) Amyloid 11:113–120.
- Sensory nerve degeneration in a mouse model mimicking early manifestations of familial amyloid polyneuropathy due to transthyretin Ala97Ser. Kan, H.W et al. (2018) Neuropathol. Appl.Neurobiol.44: 673–686.
- Amyloid deposition in a mouse model humanized at the transthyretin and retinol-binding protein 4 loci. Li X. et al. (2018) Lab.Invest.98: 512–524.
- Why is Leu55→Pro55 transthyretin variant the most amyloidogenic: Insights from molecular dynamics simulations of transthyretin monomers. Yang M. et al. (2003) Protein Science 12:1222–1231.
- The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Lashuel, H.A. et al. (1999) Biochem38(41):13560-73.
- Immunization in familial amyloidotic polyneuropathy: counteracting deposition by immunization with a Y78F TTR mutant. Terazaki H. et al. (2006) Laboratory Investigation 86: 23–31.
Leave a Reply